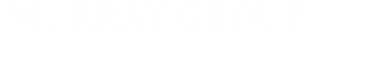
harnessing cooperative reactivity: a biological idea informing inorganic research
"nothing truly valuable can be achieved except by the unselfish cooperation of many individuals reactive centers"
our adaption of Albert Einstein
why cooperative reactivity?
Cooperative reactivity (e.g., two metal ions sharing the redox load for a reaction) is a synergistic effect that arises when multiple reactive sites work in concert or cooperate to facilitate reactions. Biological systems use cooperativity to catalyze challenging reactions including reduction of dinitrogen to ammonia, activation of strong C-H bonds, and the oxidation of water. The impressive bit isn't that these reactions can be done, but that biological systems carry out these reactions using biological reductants (e.g., NADH) under ambient conditions (e.g., temperature, pressure). If chemists develop synthetic systems with similar performance, the trickle-down societal effects can revolutionize renewable energy production, food production, and chemical synthesis.
general approach
We are synthetic inorganic chemists. In our molecular projects, we design, synthesize, and evaluate the reactivity of multimetallic complexes to understand how to control metal-ion redox cooperativity and to couple this with proximal hydrogen-bonding interactions.
------------------------------------------------------
 |
In the Haber-Bosch process, the iron-based Mittasch catalyst is the most effective for the hydrogenation of N2 to NH3, albeit at elevated temperatures and pressures. In contrast, the iron-molybdenum cofactor (FeMoco) in the active site of nitrogenase catalyzes this reduction under ambient conditions at biologically accessible potentials. Mechanistic studies on both the Mittasch catalyst and FeMoco suggest that multiple iron sites bind and activate N2. Despite the presence of multi-iron active sites in these abiological and biological systems, there are few reported attempts to replicate and control redox cooperative reactivity in synthetic iron clusters.
To probe how complex design influences reactivity towards N2, we employ macrobicyclic ligands, such as our cyclophane ligand (H3L) which contains three �-diketimine units. Our ligands define the electronic environment of each metal within the cluster and control both the distances and the electronic interactions between metal centers. Highlights from this work include the first example of a designed metal cluster capable of activating dinitrogen, and the first example of a complex containing Cu(I)�N2 interactions.
------------------------------------------------------
 |
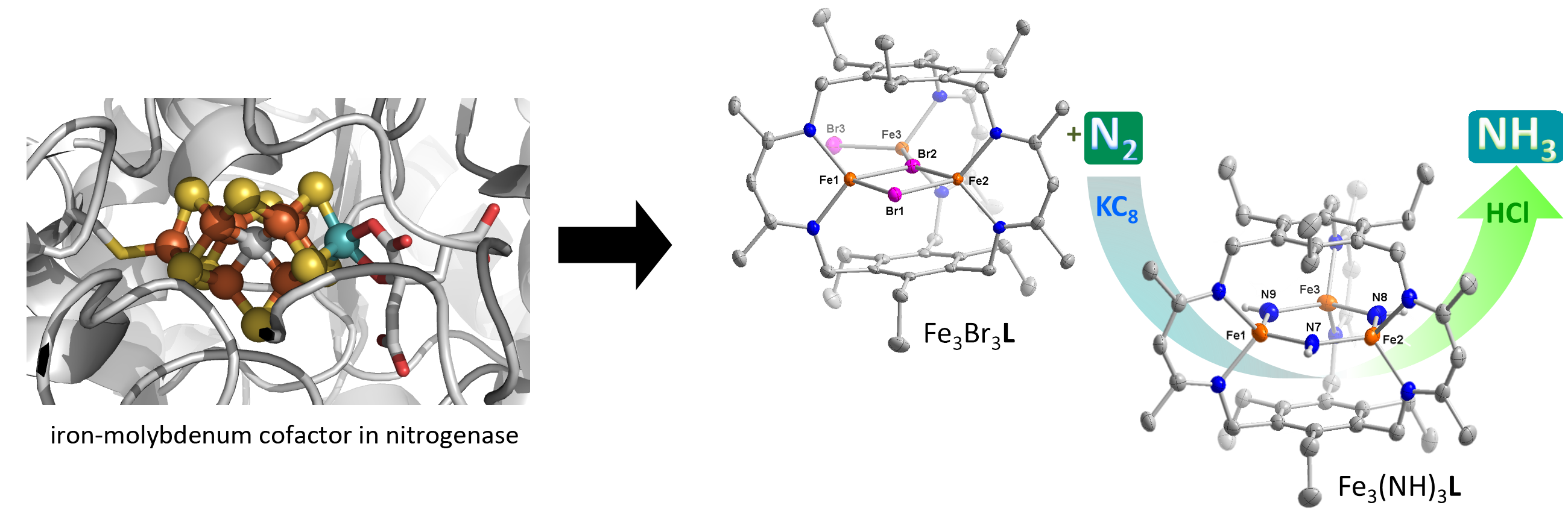
Metal hydrides have important industrial and synthetic applications, but more recently, these motifs are proposed as intermediates in enzymatic reactions, such as dinitrogen reduction in nitrogenase and H2 oxidation/H+ reduction by hydrogenases. Using our ligand design approach to control of the spatial arrangement and the electronic effects of multimetallic compounds, we want to understand and expand the chemistry of polynuclear metal-hydride clusters supported by weak-field ligands. In particular, we are interested in the parameters that partition reactivity between hydride insertion into substrates versus reductive elimination of H2.
Our group targets multinuclear 3d metal hydride clusters. For clusters housed within our cyclophane ligand, we obtain unique planar hexagonal trimetallic tri(hydride) cores. These compounds show remarkable stability towards a variety of substrates as the cluster metrics or electronic effects mute the reactivity of the hydride ligands. Highlights from this work include the first air and water stable zinc hydride, which reacts with unprecedented specificity for CO2, and the related triiron(II) and tricobalt(II) congeners, which are similarly specific for CO2.
------------------------------------------------------
 |
Copper performs diverse and essential functions throughout biology, including dioxygen transport, O2 reduction to water, N2O reduction to N2 and H2O, and CH4 oxidation to CH3OH. We are interested in how the structure of polynuclear copper compounds determines the reactivity. Although significant progress has been made in this arena, many of these systems rely on self-assembly which precludes a detailed understanding of how structure and spatial arrangement of the copper centers tune reactivity.
We focus on trinuclear copper clusters in which we can use simple reactions with atom-transfer reagents to install different bridging atoms, as well as directly probing the reaction with small molecule substrates (e.g., O2). Here, our focus is both to utilize substrate as a switch to turn-on redox cooperativity between the metal centers as well as to use single-atom bridges between the copper centers to modulate electronic communication. Highlights from work on this project include: the synthesis of the first N-ligated and coordinatively unsaturated [Cun(μn-S)] cluster, which is the closest model to the active site of nitrous oxide reductase, and our ability to selectively bind N2 in our tricopper(I)complex.
------------------------------------------------------
 |
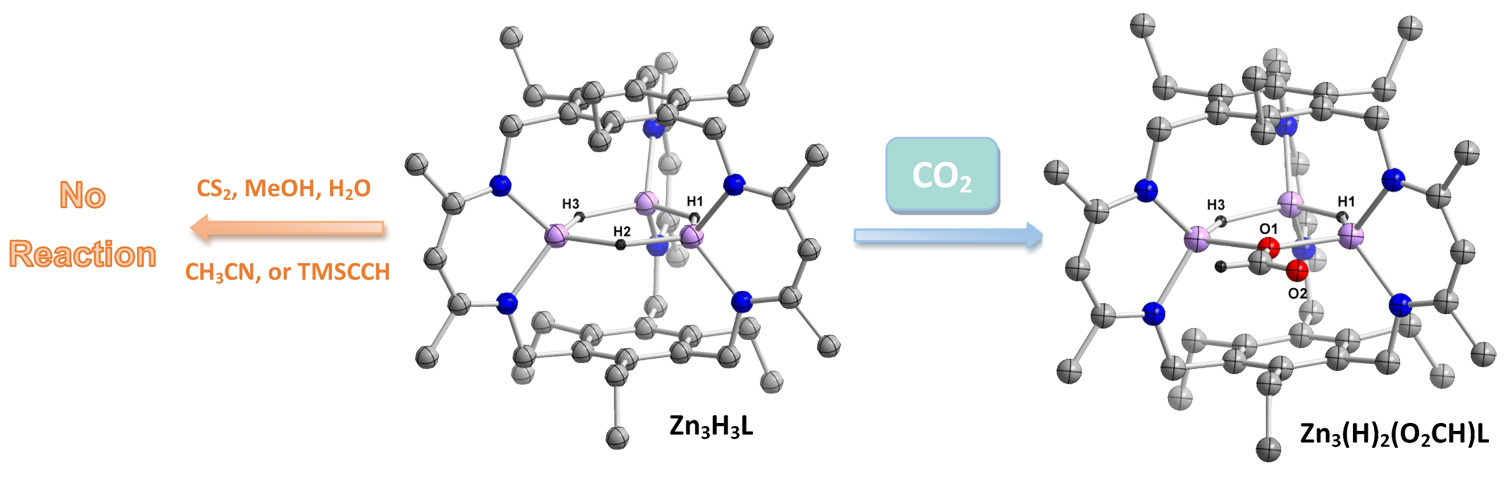
Hydrogen-bonding interactions in the secondary coordination sphere of metal centers play pivotal roles in substrate binding and activation. For example, acidic amino acid sidechains are involved in O-O bond heterolysis in cytochromes P450 as well as stabilizing the bound O2 in myoglobin and hemoglobin. We are interested in templating similar secondary coordination sphere hydrogen-bonding networks in multimetallic compounds. We envision these interactions can deliver proton equivalents during the course of chemical reactions or orient substrates in the appropriate manner.
Our approach is to develop facile routes to selectively and partially metalate macrobicyclic ligands. Given our typical choice of metal-binding groups (e.g., pyridine dicarboxamides or diketimines), these partially-metalated complexes are anticipated to template hydrogen-bond networks within the ligand cavity. We have demonstrated proof of concept with our discovery of a simple route to dimetallic complexes of our cryptand ligand. Indeed, hydrogen-bonding interactions are incorporated in the secondary coordination sphere of these dicopper and dinickel cryptate complexes.
 |
University of Florida-
Department of Chemistry PO Box 117200, Gainesville, FL 32611-7200 |